


A kergemarhakór járványszerű elterjedése Nagy-Britanniában ráirányította a közfigyelmet az ún. prionbetegségekre, amelyekhez olyan szindrómák tartoznak, mint pl. a Creutzfeldt–Jacob-betegség vagy a Pápua Új Guineán korábban elterjedt nevetőhalál. Ezek a fertőző betegségek azzal az egyedülálló tulajdonsággal rendelkeznek, hogy kialakulásukat nem hagyományos kórokozók, tehát baktériumok, vírusok vagy gombák, hanem speciális tulajdonságú fehérjék okozzák, amelyek a szervezetbe jutva egészséges fehérjék szerkezetét módosítják. Jelen írás áttekinti a fehérjéket, a fehérjék térszerkezeti hibáit, illetve az ezekből eredő – egyelőre sajnos még gyógyíthatatlan – betegségeket (pl. Alzheimer-, Parkinson-kór), hogy a fertőző prionbetegségek sajátságai értelmezhetővé váljanak. A cikk a fehérjeszerkezet alapelvein keresztül magyarázza a betegségek kialakulásának molekuláris okait, és röviden tárgyalja a beavatkozások lehetséges jövőbeni módozatait. Továbbá érdekes kitekintést ad arra, hogy a szerkezet sokszor betegséget okozó változásai a fehérjék mindennapi működésében is fontos szerepet játszhatnak, vagyis részét képezik sejtjeink funkcionális fegyvertárának.
A fehérjeszerkezet alapjai
Legtöbbször nem vagyunk tudatában annak, hogy az életünkben milyen meghatározó szerepet játszanak a fehérjék. Gyakran csak a táplálék részének tekintjük őket: tudjuk, hogy fontos a fehérjében gazdag étrend, de hogy pontosan miért is, arra valószínűleg kevesen ismerik a választ. Pedig a fehérjék szervezetünk meghatározó építőelemei, kis túlzással azt mondhatnánk, az egész szervezetünk fehérjemolekulák halmaza, és egész életünk nem más, mint fehérjemolekulák bonyolult, összehangolt tevékenysége, működése. Fehérjék végzik az ételben található tápanyagok lebontását, majd a lebontás során keletkező kis molekulákból szervezetünk saját anyagainak – többek között fehérjéinek – felépítését is. Fehérjék másolják örökítő anyagunkat, a DNS-t, közömbösítik a szervezetünkbe kerülő ellenséges mikroorganizmusokat, szállítják vérünkben az oxigént, és teszik lehetővé, hogy izmaink összehúzódjanak. Teszik a dolgukat anélkül, hogy tudomást vennénk róluk, egészen addig, amíg valamelyik meg nem sérül, el nem romlik, enyhébb vagy éppen súlyosabb betegséget okozva. Ilyenkor megtanuljuk annak az egynek a nevét, amelyik a bajt okozta.
A humángenom-projekt sikeres befejezése óta tudjuk, hogy kromoszómáinkban összesen mintegy 30 ezer gén található. A gének az öröklődés alapegységei, feladatuk általában az, hogy egy fehérje létrehozásához szükséges információt kódoljanak. Ezen egyszerű megfeleltetés alapján – némi egyszerűsítéssel – azt mondhatjuk, hogy szervezetünk 30 ezer alapvetően különböző fehérjét képes előállítani. A teljes igazság persze ennél egy kicsit bonyolultabb, mivel az egyes fehérjék szintézise még nagyon sok egyedi variációt is megenged, így az elvileg ténylegesen létrejöhető fehérjék száma közel egymillió. A fehérjék kémiai szempontból polipeptidek, vagyis aminosavak egymásután kapcsolódásával létrejövő hosszú láncok (1. ábra). Az emberi szervezet – és minden más ismert életforma – fehérjéit mindössze húsz aminosav alkotja, viszont az aminosavak kapcsolódási sorrendje a fehérjében gyakorlatilag tetszőleges, és ez az, amiben az egyes fehérjék eltérnek egymástól. A helyzet nagyon hasonló az ember által beszélt nyelvekre, ahol kis számú betű kombinálásával nagyon sokféle szó és szinte végtelenül sokféle mondat rakható össze. Egy átlagos fehérje hossza mintegy 300 aminosav, így kémiailag felfoghatatlanul sokféle (20300) fehérje jöhet létre, aminek a valóságos fehérjék csak a törtrészét teszik ki.
Logikus a kérdés, hogy a szervezetünkben található fehérjék a csaknem végtelen lehetőség közül valamilyen szempontból kitüntetettek-e, vagy éppenséggel teljesen véletlenszerűek. A biokémiai/fehérjekémiai kutatások elmúlt évtizedeinek egyik legfontosabb felismerése, hogy a fehérjék mind rendelkeznek egy speciális tulajdonsággal, amennyiben képesek egy jól meghatározott térbeli elrendeződést fölvenni. Valahogy úgy, ahogy egy fonalat gombolyaggá tekerünk, a sejtben történő szintézisét követően a polipeptidlánc is felgombolyodik, és az aminosavak közötti kedvező kölcsönhatások, vonzóerők következtében jellegzetes térszerkezetet vesz fel (1. ábra). Ezek a kölcsönhatások az aminosavak fizikokémiai tulajdonságaiból következnek; megkülönböztetünk van der Waals, hidrogénhidas és ionos kölcsönhatásokat. A nagyszámú gyenge kölcsönhatás által stabilizált térszerkezet minden egyes fehérje esetében más és más, ez teszi lehetővé a fehérje működését, funkciójának ellátását. A térszerkezetben az atomok pontos elhelyezkedését speciális „fényképezési” eljárásokkal (röntgen krisztallográfia, NMR) meg tudjuk határozni, és így a fehérjeszerkezet-funkció összefüggéseiről pontos képet tudunk alkotni.
A stabil térszerkezet kialakításának képessége természetesen nem magától értetődő és nem is véletlen tulajdonság, hanem hosszú fejlődési folyamat eredménye. Ennek alapja, hogy az aminosav-sorrendet meghatározó genetikai állomány másolása és generációról generációra való átadása, vagyis az öröklődés, nem teljesen pontos folyamat. Valahogy úgy, ahogy a kódexmásolók időnként hibákat vétettek, és ezért a kódexek idővel változtak, a génekben is változások, mutációk léptek – és lépnek a mai napig is – fel. Ennek következtében az általuk kódolt fehérjék aminosav-sorrendje is változatosságot mutat, vagyis egy adott fehérjének mindig sokféle változata keletkezett és működött különféle fajokban és azok egyedeiben. A sokféle változat közül a funkció szempontjából hatékonyabbak, vagyis szerkezetileg stabilabbak előnyt jelentettek a szervezet számára, és így az evolúció során kiszelektálódtak. Ez a lassú, évmilliárdokon keresztül tartó folyamat válogatta ki azokat a fehérjéket, amelyek aminosav-sorrendje stabil térszerkezet kialakítását teszi lehetővé.
A folyamat természetesen még ma sem ért véget, amit a mutációk, egyedi variánsok, polimorfizmusok gyakori volta és maguk a fehérjeszerkezet hibáira visszavezethető betegségek is mutatnak. A fehérjék térszerkezetével kapcsolatosan egy további tudnivaló, hogy a lánc feltekeredése gyakran nem megy magától, mivel a sejtek belsejében nagyon nagy számú más fehérje is található, ami rendkívül megnöveli a fehérjék összetapadásának, így működésképtelen és sok esetben káros aggregátumok képződésének a veszélyét. Ezek kialakulása ellen fehérjék két speciális csoportja véd bennünket. Az egyik csoportba a dajkafehérjék (chaperonok) tartoznak, amelyek átmenetileg az újonnan szintetizálódott polipeptidlánchoz kapcsolódnak, hogy megvédjék az egyéb fehérjékkel való érintkezéstől, és elősegítsék ez által a helyes térszerkezet kialakulását. A másik csoportot olyan fehérjék (ubikvitin konjugáló enzimek, proteaszóma) alkotják, amelyek felismerik a helytelenül feltekeredett, hibás térszerkezetű fehérjét, és alkotóelemeire, vagyis aminosavakra bontják le.
A funkciót hordozó térszerkezet kialakulása tehát a szervezet „boszorkánykonyhájában” folyó, alaposan felügyelt, de nagyon bonyolult folyamat, amelybe néha hibák csúsznak. Ezek a hibák persze hosszú távon, az evolúció időskáláján nyersanyagot biztosítanak a hatékonyabb változatok kikísérletezéséhez, de az egyén vagy egyed szintjén súlyos betegségekként nyilvánulhatnak meg.
Hibák a szerkezetben
A fehérjeszerkezet hibái gyakran arra vezethetők vissza, hogy a polipeptidlánc nemcsak egy térszerkezetet vehet fel, hanem van egy olyan térbeli elrendeződése is, ami az aktuális aminosav-sorrendtől függetlenül stabil állapotot tesz lehetővé. Mint korábban elhangzott, a funkcionális térszerkezet kialakulása az aminosavak közötti nagyszámú kedvező kölcsönhatás eredménye. Két párhuzamosan futó polipeptidlánc között azonban a konkrét aminosav-sorrendtől függetlenül is nagyszámú hidrogénhíd alakulhat ki: ez az ún. béta-szerkezet a láncban található aminosavaktól függetlenül stabil, ezért kialakulásával gyakorlatilag minden fehérje esetében számolni kell. A béta-szerkezet geometriájából következően a párhuzamosan futó lánco(ka)t egy irányba akárhányszor meg lehet ismételni, amiből következően az a fehérje, ami valamilyen okból az ilyen szerkezet kialakulásának útjára lépett, gyakorlatilag visszafordíthatatlanul nagyméretű, gyakran szabad szemmel is látható, sok fehérjemolekulát magában foglaló aggregátumot alkot (2. ábra). Ezt az aggregátumot amiloidnak hívjuk, és a létrejöttével kapcsolatosan az a legnagyobb probléma, hogy a szervezet molekuláris védekező mechanizmusai hatástalanok vele szemben, sem a dajkafehérjék, sem a lebontó proteaszóma nem képes hozzáférni. Kialakulása ezért gyakran irreverzíbilis, visszafordíthatatlan. Figyelembe véve a sejtekben található hatalmas fehérjekoncentrációt és az amiloid stabilitását, a védekező és biztosító mechanizmusokra, illetve a fehérjék feltekeredésének hatékonyságára vonatkozóan is igen nagy eredmény, hogy a fehérjeszerkezet hibái, az amiloidózisok viszonylag ritkán következnek be.
Az amiloid kialakulásához kapcsolható betegségek általában idősebb korban jelentkeznek, és alapvetően három okra vezethetők vissza. Mindhárom ok nem megfelelő térszerkezetű, félig feltekeredett fehérjemolekulák (2. ábra) felhalmozódását idézi elő, ami ezek összetapadását és így az amiloid kialakulását váltja ki. Gyakran érheti a fehérjéket mutáció, vagyis aminosav-sorrendjük egy helyen történő véletlen megváltozása, amely destabilizálja a fehérje térszerkezetét, és ezen keresztül idéz elő aggregációt. Mivel maga a betegség gyakran csak idős korban jelentkezik, addigra a mutáció az utódokra is továbbadódhat, és így örökletessé válik. A fehérjeszerkezetet nagyobb mértékben destabilizáló mutációk ugyanakkor már fiatal korban is előidézhetnek amiloidózist, ami ilyen esetben súlyosabb következménnyel járó, gyorsabb lefolyású betegséget okoz. Bonyolítja persze a helyzetet, hogy az előbbiek értelmében a fehérje térszerkezetének kialakulásában és működésében sok más fehérje is részt vesz, így az amiloidózis nem csak az adott fehérjét, hanem más fehérjét érintő mutációk esetében is kialakulhat, vagyis a betegség poligénikus lehet.
A betegségek kialakulásának másik lehetséges útja, hogy a normál aminosav-sorrendű – vagyis mutációt nem szenvedett – fehérje bizonyos valószínűséggel elveszíti stabil térszerkezetét, letekeredik, és a javító/lebontó mechanizmusokat elkerülve aggregál. A kezdeti aggregátumot azután, az aggregátum stabilitása és ellenálló képessége miatt, a szervezet már nem tudja eltávolítani, tartós jelenléte további aggregációhoz és végül a betegség kialakulásához vezet. Ezeknek a szórványos vagy sporadikus formáknak a megjelenése kizárólag idős korban történik, mivel a kezdeti fehérjeaggregátum normál fehérjéből történő kialakulása energetikailag nagyon kedvezőtlen, lassú folyamat, és csak a javító mechanizmusok csökkent hatékonysága, elöregedése mellett történhet meg.
A harmadik kiváltó ok tulajdonképpen az előző kettőből kiindulva különleges körülmények következménye, amennyiben bizonyos fehérjeaggregátumok azzal a speciális tulajdonsággal rendelkeznek, hogy egy másik szervezetbe jutva képesek az ott található, egészséges fehérjéket is az aggregáció útjára kényszeríteni. Az ilyen, megváltozott térszerkezetű fehérjék ezért fertőzőek; ezt a különleges fehérjeosztályt prionnak nevezzük. Különleges szerkezeti tulajdonságától eltekintve a prion normális élettani funkciót ellátó fehérje.
A betegségben megnyilvánuló tünetek kialakulásáért alapvetően kétféle molekuláris mechanizmus tehető felelőssé, amelyek a különböző esetekben eltérő súllyal esnek latba. Az egyik lehetőség, hogy a hibás térszerkezetű fehérje nem képes korábbi funkcióját ellátni, vagyis az amiloid kialakulása funkcióvesztéssel párosul. Mivel a fehérjék térszerkezete a működéshez nélkülözhetetlen, és az amiloidózisokban szerepet játszó fehérjék a szervezet számára fontos funkciókat látnak el, reális ennek a mechanizmusnak a jelentősége. Szervezetünk minden génből két példányt (allélt) hordoz, vagyis minden fehérje szintézise két gén információi alapján folyik, ezért a funkcióvesztéses mutáció jellegzetessége, hogy recesszív módon öröklődik. Ez annyit jelent, hogy a tünetek nem jelentkeznek, ha a mutáció csak az egyik allélt érinti, mivel a másik allél irányításával szintetizálódó fehérje képes a funkciót ellátni. Nem ez a helyzet a másik esetben, amikor is a hibás térszerkezet kialakulása új funkció kialakulásával, funkciónyeréssel párosul. Az amiloid képes lehet például más, kis mennyiségben jelen lévő fehérjék (pl. a gének átíródását szabályozó transzkripciós faktorok vagy a sejtek kommunikációjában meghatározó szerepet játszó receptorok) megkötésére és gátlására. Ebben az esetben a betegséget elsősorban ez az új hatás (funkció) okozza, és nem az amiloidózisért felelős fehérje működésének kiesése. Erre a mechanizmusra utal a betegség domináns volta, vagyis annak ellenére történő kialakulása, hogy az amiloidózisért felelős mutáció csak az egyik allélt érinti, így a szervezetben az amiloidot alkotó fehérje normál térszerkezetű, funkcióképes változata is jelen van.
Idegölő hibák
Az amiloidózisok legtitokzatosabb és egyben legfélelmetesebb változatai a neurodegeneratív fehérjeszerkezeti hibák, amelyek szövet-specifikus fehérje-lerakódással járnak együtt, amelyek általában az agyat érintik, és súlyos idegsejt-pusztulást okoznak (1. táblázat). Bár az egyes betegségek, mint az Alzheimer-betegség, Parkinson-kór, Huntington-betegség, valamint a prionbetegségek mögött más-más fehérje hibája húzódik meg, a közös tárgyalást a hasonló patomechanizmus (a fehérjeszerkezet hibája által okozott amiloidózis és az amiloid okozta idegsejt-pusztulás), valamint a hasonló tünetegyüttes (fokozatos rosszabbodás [progresszivitás], elbutulás [demencia], érzelmi/pszichés zavarok és mozgáskoordinációs problémák, később teljes szellemi leépülés) indokolja. További közös pont, hogy a betegségek gyógyítására egyelőre nem vagyunk képesek, csupán tüneti kezelésükre van mód bizonyos esetekben.
1. táblázat. A leggyakoribb neurodegeneratív fehérjeszerkezeti betegségek
Betegség Előfordulás* Aggregátum Gén/fehérje
Alzheimer 4 millió plakk és fonadék APP, PS1, PS2
Parkinson 1 millió Lewy-test alfa-szinuklein, parkin, UCHL1
Huntington 30 ezer huntingtin-inklúzió huntingtin
CJD 400 prion-aggregátum PrP
*az USA-ban
Alzheimer-betegség
Az Alzheimer-kór tipikus időskori betegség, egyben a legelterjedtebb amiloidózis és az egyik legelsőként leírt fehérjeszerkezeti betegség. Alois Alzheimer írta le 1907-ben, hogy az általa kezelt, bizonyos tünetekkel elhunyt betegek agyában az agykéreg idegsejtjeinek felületén fehéres lerakódások, plakkok figyelhetők meg. Későbbi vizsgálatok azonosították, hogy ezek fő komponense a sejtfelszíni amiloid prekurzor fehérje (APP) egy rövid darabja. A betegek agyában a sejtek belsejében is kimutatható a szokatlan fehérjeaggregátumok, a helikális filamentumok jelenléte, amelyek fő alkotóeleme egy másik fehérje, a tau. A betegség elsődleges oka azonban a plakkok kialakulása, amit ritkábban mutációk (5%), sokkal gyakrabban ismeretlen környezeti hatások és véletlen, sporadikus fehérjeszerkezeti változások idéznek elő. Az Alzheimer-betegség enyhébb memóriazavarral, idő- és térbeli tájékozódási problémákkal, a kezdeményező készség csökkenésével kezdődik. A középső stádiumban már jelentős emlékezetzavar lép fel, a beteg a családtagok nevét és hétköznapi szavakat sem tud felidézni, a mindennapi tevékenységben (öltözködés, tisztálkodás) is segítségre szorul. Hallucinációk, érzelemkitörések, depresszió nehezíti a helyzetet. A végső stádiumra a személyiség teljes leépülése jellemző, a beteg gyakorlatilag senkit nem ismer fel, és állandó felügyeletre szorul. A fokozatos romlás végül is átlagosan 7-10 év alatt vezet halálhoz. Az örökletes formáktól eltekintve a kiváltó ok(ok) nem ismert(ek), gyógymódja nincs. Tüneti kezelésére alkalmazhatók a lelki problémákat (depresszió, hallucinációk) enyhítő gyógyszerek, az általános agyi anyagcserét, vérellátást javító szerek, illetve újabban az agy egyik fontos információközvetítő (neurotranszmitter) molekulája, az acetilkolin szintjének emelkedését előidéző készítmények.
Parkinson-kór
A Parkinson-kór a második leggyakoribb fehérjeszerkezeti neurodegeneratív betegség, James Parkinson írta le először 1817-ben. Kiváltó oka egy ismeretlen funkciójú fehérje, az alfa-szinuklein aggregációja, amit az esetek kisebb hányadában itt is mutáció okoz. A betegség kialakulásában az oxidatív stressz és bizonyos környezeti faktorok (rovarirtók, permetezőszerek), valamint két további gén mutációja is szerepet játszhat. A kialakuló aggregátumokat Lewy-testeknek hívjuk, és elsősorban az agy dopaminerg (dopamin neurotranszmitterrel működő) idegsejtjeiben fordulnak elő. A Lewy-testek toxicitása a dopaminerg idegsejtek pusztulását váltja ki, ami a fontos információközvetítő molekula egyensúlyának felborulásához és ezen keresztül mozgási zavarokhoz (remegés, izommerevség, mozgások lassúsága, mozgásszegénység) vezet. A fizikai tünetek mellett a Parkinson-kórnál is felismerhetők pszichés problémák (szorongás, depresszió), majd a későbbi fázisban az elbutulás. A Parkinson-kór fontos abból a szempontból, hogy mivel pontosan ismert, hogy a betegség tüneteit a dopamin hiánya okozza, a dopaminszintet helyreállító gyógyszerekkel lehetőség van tüneti kezelésére. Ezek a gyógyszerek általában vagy a dopamin termelődését segítik elő (pl. a dopamin előalakja, L-dopa), vagy a lebomlását gátolják (antagonisták, pl. MAO-B inhibítorok). Az aggregáció vagy az idegsejtek pusztulásának visszafordítása azonban mai ismereteink szerint nem lehetséges, bár őssejtek beültetésével folynak már állatkísérletek.
Huntington-betegség
A Huntington-betegség jóval kevésbé elterjedt, mint az Alzheimer- vagy a Parkinson-kór, fontosságát azonban az adja, hogy egy szokatlan fehérjeaggregációs betegségcsoport, a poliglutamin- (vagy CAG repeat) betegségek legjobban ismert és jellemzett képviselője. Az előzőektől eltérően ezen betegségek kiváltó oka mindig egy adott fehérje mutációja, mivel a betegségek arra vezethetők vissza, hogy egy neuronális fehérjében (a Huntington-kór esetében a huntingtinben) a glutamin nevű aminosav (Q) a polipeptidláncban egymás után nagyon sokszor megismétlődik. A fehérje adott szakasza egészséges egyénekben is a glutamin ismétlődését (Q15–Q36) mutatja, ami azonban még nem okoz problémát. A normális repetíció viszont veszélyt hordoz magában, mivel az ilyen ismétlődő szekvenciarészleteket hordozó gének általában nem stabilak, másolódásukkor gyakran hibák történnek, ami az ismétlődések számának megnövekedésében nyilvánul meg. Ez a mutáció igen gyakori, egyes egyénekben a glutamin ismétlődési száma a 120-at is elérheti, ami a fehérje oldhatóságának komoly csökkenésével jár. Ez aggregátumok kialakulásához vezet. A Huntington-betegség esetében gyakori jelenség, hogy a glutamin ismétlődési száma generációról generációra növekszik, és ezzel párhuzamosan a betegség egyre fiatalabb korban jelentkezik. Az aggregátumok a striátumban és egyéb agykérgi területeken az idegsejtek belsejében jelennek meg, és a sejtek halálához vezetnek. A Huntington-kór leggyakrabban 35-40 éves korban mozgáskoordinációs zavarokkal, akaratlan, rángásos mozdulatokkal kezdődik, személyiségváltozásokkal, elbutulással, a mozdulatok vitustáncig való fokozódásával folytatódik, és általában 15 év alatt halálhoz vezet.
Prionok
A legrégebben – több mint 200 éve – ismert prionbetegség a juhokat érintő scrapie (surlókór). Egy másik, emberekre veszélyes betegség, a kuru (nevetőhalál) járványszerűen terjedt Pápua Új Guineán, és a XX. század első felében szedte áldozatait. A betegséget a bennszülöttek rituális kannibalizmusa, főképp az elhunyt rokonaik agyvelejének elfogyasztása okozta, nevét a végső stádiumban vigyorszerű grimaszra torzult arcról és a ziháló, nevetésszerű lélegzésről kapta. Bár a betegség terjedésének mechanizmusát akkor még nem ismerték fel, fertőzés tényére utalt, hogy az esetek száma csökkenni kezdett, amikor a hittérítők a barbár szokást betiltották. Daniel C. Gajdusek, aki elsőként vizsgálta a betegséget, a fertőzést többéves lappangási ideje miatt egy új típusú patogén, a lassú vírus hatásának tulajdonította. A prionbetegség egy további ismert válfaja a Creutzfeldt–Jakob-betegség (CJD), amely gyakran örökletes módon jelenik meg, időskori személyiségváltozást, kognitív problémákat és koordinációs zavarokat okoz. A további emberi prionbetegségek (halálos családi álmatlanság, Gerstmann–Sträussler–Scheinker-tünetegyüttes) meglehetősen ritkák, és általában a nagyközönség számára kevéssé ismertek.
A közfigyelmet a problémakörre végül is prionbetegségek legújabb válfaja, a kergemarhakór vagy szarvasmarha szivacsos agyvelőgyulladás/sorvadás (Bovine Spongiform Encephalopathy, BSE) irányította. Ez a betegség kényszerítette ki a nagy-britanniai szarvasmarha-állomány kiirtását, mivel felmerült a gyanú, amely később be is igazolódott, hogy a CJD egy fiatalkorban megjelenő, új variánsának (vCJD) megjelenéséért a fertőzött, de a betegség tüneteit még nem mutató állatok húsának fogyasztása a felelős. A vCJD áldozatainak száma a mai napig Nagy-Britanniában mindössze 140, de a legújabb, különböző műtétek során eltávolított szövetminták elemzésén alapuló átfogó vizsgálat szerint a prionnal akár több ezren is fertőzöttek lehetnek. A BSE járványszerű elterjedését valószínűleg az okozta, hogy a szarvasmarhákat állati maradványokból készített takarmánnyal etették. E gyakorlat megszüntetése után – a kuruhoz hasonlóan – a BSE-esetek száma csökkenni kezdett (3. ábra), az állomány kiirtását azonban az emberre való átterjedés indokolta.
A prion egyedülálló viselkedésének magyarázatát elsősorban két kiemelkedő kutató, D. C. Gajdusek és Stanley B. Prusiner munkásságának köszönhetjük, akik vizsgálataikért Nobel-díjban részesültek. Gajdusek volt az első, aki felismerte az egymástól látszólag független betegségek (scrapie, kuru, CJD) kialakulásának és lefolyásának hasonlóságait, és így rámutatott, hogy ezek valószínűleg hasonló okra visszavezethető neurodegeneratív betegségek. Megfigyelései indították el az akkor még lassú vírusnak gondolt kórokozó keresésére irányuló kutatásokat. A lassú vírus vagy más hagyományos kórokozó (baktérium, gomba) megtalálására irányuló erőfeszítések azonban rendre kudarcot vallottak, és a rejtély megoldása csak Prusiner színrelépésével került elérhető közelségbe.
Prusiner kezdeti megfigyelései a kórokozó szokatlan viselkedését mutatták, mivel azt találta, hogy ellenáll minden olyan kezelésnek (UV-besugárzás, DNS-emésztés, hőkezelés, kémiai sterilizáció), amely a hagyományos kórokozókat tönkreteszi. A pontos azonosításban áttörést jelentett, amikor a BSE-t és a CJD-t kínai aranyhörcsögökre sikerült átvinnie, mivel ezekben az állatokban a fertőzést követően a betegség viszonylag hamar (mintegy fél év alatt) kialakul, így lehetőség nyílt a kórokozó izolálására és azonosítására. Nagy meglepetésre a jelentősen megtisztított, fertőzőképes preparátumok csak fehérjét tartalmaztak, hagyományos értelemben vett örökítő anyagot (DNS, RNS) nem. Még nagyobb volt a meglepetés, amikor kiderült, hogy ez a fehérje az egészséges állatokban és emberekben is előfordul, vagyis szervezetünk mindennapos működésében is szerepet játszik. Ilyen megfigyelések alapján fogalmazta meg Prusiner „csak fehérje” (protein only) hipotézisét, amely szerint a fertőzésért, a betegség terjedéséért nem egy hagyományos kórokozó, hanem egy hibás szerkezetű fehérje a felelős, amely a normális térszerkezetű, egyébként azonos fehérje szerkezetét megváltoztatja, és a maga képére formálja. A kezdeti szerkezetváltozás a többi betegséghez hasonlóan a prion esetében is lehet mutáció vagy véletlen átalakulás következménye. Egyedien jellemző azonban erre a betegségre a fertőzőképesség, vagyis hogy a prion kívülről, nagyon kis mennyiségben a szervezetbe jutva is képes kiváltani a betegséget. A fertőzés lehet táplálkozás (állattáp vagy állati eredetű ételek fogyasztása) vagy orvosi beavatkozás (szaruhártya-átültetés, fertőzött elektróda beültetése, növekedésihormon-készítmény adása) következménye. Mindezen körülmények indokolták, hogy a kórokozó elnevezésére új kifejezést alkossanak, ez a fehérjetermészetű fertőző részecske angol megfelelőjének (proteinaceous infectious particle) rövidítéseként prion lett.
Ellenségből barát?
A prion fertőzőképessége egy érdekes és izgalmas lehetőséget is felvet. Mivel a prion megváltozott térszerkezete a többi prionmolekulának is átadódik, ez a fehérjemolekula olyan információhordozónak tekinthető, amelyik a változást kiváltó külső körülményre, környezeti hatásra elvileg akármeddig „emlékezik”. Más szavakkal, a prion szerkezete olyan emléknyomként működhet, amelyik megfelelő körülmények között az élőlényre kedvező, tartós információmorzsát hordozhat, aminek az alkalmazkodásban, a környezetre való megfelelő reagálásban szerepe lehet.
Alapvetően az élőlények kétféleképpen tárolhatnak információt. Az egyik mód a genetikai információtárolás, amely a DNS bázissorrendjébe írva generációról generációra öröklődő módon tartalmazza az élőlény felépítésére, általános működési szabályaira vonatkozó utasításokat. Ez határozza meg az egyes fajok egyedi jellegzetességeit, és a tartalma csak nagyon lassan, evolúciós időskálán változik. A másik nagy információhordozó mechanizmus az élőlény élete alatt a külvilágról szerzett információk tárolásának és megfelelő módon való visszahívásának képessége, amit bizonyos esetekben memóriának nevezünk. Az így tárolt információ az öröklődésben elvész, vagyis utódainknak közvetlenül nem adódik tovább, viszont ez biztosítja, hogy a genetikai információ által meghatározott kereteken belül az élőlény a környezetére minél hatékonyabban legyen képes reagálni. Mindkét információtárolási képesség alapvetően fontos az élőlény működése szempontjából.
A priont szerkezeti sajátossága elvileg alkalmassá teszi arra, hogy megfelelő molekuláris kontextusba illesztve a szerzett információ tartós hordozója legyen. Ez a felismerés vezetett néhány éve annak a hipotézisnek a megfogalmazásához, hogy a priont ne csak betegséget okozó fehérjének, hanem az élőlény számára hasznos információhordozónak, esetleg memóriamolekulának tekintsük. A hipotézis ellenőrzését kezdetben élesztősejteken végezték, ahol sikerült is kimutatni, hogy bizonyos fehérjék képesek prion-szerű szerkezetváltozásokra, és megváltozott szerkezetük a sejt túlélése szempontjából hasznos lehet. Konkrétan, az Ure2 fehérje szerkezetileg átrendeződhet olyan módon, hogy a vele kapcsolatban lévő fehérjéket is befolyásolva képessé tegye az élesztősejtet nitrogénmentes környezetben egy adott nitrogénforrás (urea) hasznosítására. Bár ez nem tűnik túl jelentős haszonnak, az élesztősejtek számára az evolúciós versenyben bizonyos körülmények között a túlélést biztosíthatja.
A memóriafunkcióval kapcsolatban még jelentősebb felismerés fűződik Erik Kandel nevéhez, aki az állati memóriajelenségek kutatásában elért eredményeiért nemrég Nobel-díjat kapott. Kandel vizsgálatainak középpontjában egy tengeri csiga (Aplysia californica) áll, amely viszonylag egyszerű idegrendszerrel és jól reprodukálható memóriajelenségekkel, pl. egyszerűen kialakítható és vizsgálható reflexekkel rendelkezik. A tengeri csiga memóriájának molekuláris szintű vizsgálata azt mutatta, hogy a memória az idegsejtek kommunikációjának tartós megváltozásában „kódolódik”, ami a bennük található fehérjemolekulák szintjének, eloszlásának, aktivitásának megváltozására vezethető vissza. Az egyik ilyen fehérjemolekuláról (CPEB, citoplazmatikus poliadenilációs szignálkötő fehérje) az elmúlt év során kiderült, hogy priontulajdonságokkal rendelkezik, vagyis szerkezetének tartós megváltoztatására és a változás továbbadására képes, továbbá, hogy az idegsejtek kommunikációjában játszik szerepet. Ez a prion a gazdaszervezet számára semmilyen veszélyt nem jelent, épp ellenkezőleg, priontermészete az élőlény túlélési és szaporodási, vagyis összességében evolúciós esélyeit javítja.
Javíthatatlanok?
A fehérjeszerkezeti betegségek e pillanatban nem gyógyíthatók, felismerésüket követően a beteg állapotában lassú, de biztos romlás következik be. Legjobb a helyzet a Parkinson-kór esetében, amely a lokális idegsejt-pusztulás következtében jól körülírható tünetekkel jár együtt, és tüneti kezelésére a fent említett módon jó lehetőség van. Újabban bizonyos próbálkozásokban az idegsejtek pótlására tesznek kísérletet, ami valószínűleg tényleges áttörést hozhat a nem túl távoli jövőben. A többi betegségnél a tüneti kezelésre is alig van lehetőség, az egyetlen jelenlegi reményt a születés előtti (prenatális) diagnosztika nyújtja, amellyel az örökletes formák esetében elvileg már korai magzati korban lehetőség van a betegség felismerésére és kiszűrésére. Sajnos ezek a formák a betegségeknek csak kis hányadát teszik ki, és mivel a betegségeket több fehérje mutációja is kiválthatja, a felismerés nem mindig egyértelmű.
Kísérleteznek olyan gyógyszerekkel is, amelyek a fehérjeszerkezet kedvezőtlen átrendeződésébe avatkoznak be. Ez elvileg két úton lehetséges, szóba jöhetnek a normális (natív vagy fiziológiás) térszerkezethez kötődő, azt stabilizáló gyógyszerjelöltek, vagy éppen a megváltozott térszerkezetű fehérjemolekulák egymáshoz tapadását, az amiloid kialakulását gátló molekulák. Elképzelhető ugyanakkor harmadik út is. Említettem, hogy a szerkezeti betegségek kialakulásában a fehérjéket gondozó sejtes rendszerek (dajkafehérjék, lebontási rendszer) elöregedése, „kifáradása” is szerepet játszik, hiszen ennek következménye, hogy az örökletes formák, amelyekben a mutációt az egyén egész életében hordozza, gyakran csak idősebb korban manifesztálódnak. Ez felveti azt a lehetőséget, hogy a betegségek megelőzhetők, illetve kezelhetők lesznek olyan gyógyszerekkel, amelyek ezen rendszerek hatékonyságát növelik. A gyógyszergyárakban természetesen ilyen irányú fejlesztések napjainkban is folynak.
Természetesen ezzel párhuzamosan, nagy erőkkel folynak tovább a kutatások a világ akadémiai kutatóintézeteiben és gyógyszergyáraiban, hogy a fehérjék térszerkezeti változásainak okait felderítsék. Reményeink szerint ezek eredményei nem csak a szörnyű betegségekbe való beavatkozás lehetőségét teremtik meg, hanem általuk közelebb kerülhetünk agyunk legmagasabb rendű funkcióinak, személyiségünk legbelső titkainak molekuláris szintű megértéséhez is.
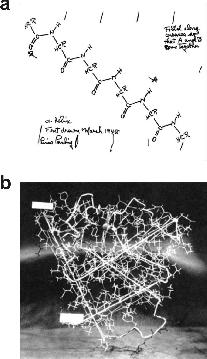
1. ábra. A fehérjék kémiai szerkezete és térszerkezete
A fehérjék kémiai szempontból polipeptidek, vagyis aminosavak egymásután kapcsolódásával kialakuló hosszú láncok. A polipeptidlánc általános szerkezeti sémáját Linus Pauling, a kétszeres Nobel-díjas kutató eredeti rajza mutatja, aki ezen a vázlaton tette közzé először az amiloidózisban is kulcsszerepet játszó nyújtott béta-szerkezettel kapcsolatos elképzeléseit (a). A polipeptidlánc feltekeredése és az aminosavak közötti nagyszámú kedvező kölcsönhatás következtében a fehérje egyedi térszerkezetet vesz fel, amelyben minden atom az adott fehérjére jellemző térbeli pozíciót foglal el (b). A szerkezeti séma a John Kendrew és Max Perutz által röntgenkrisztallográfia segítségével meghatározott első fehérjeszerkezetet, az oxigéntároló mioglobin térszerkezetét mutatja.
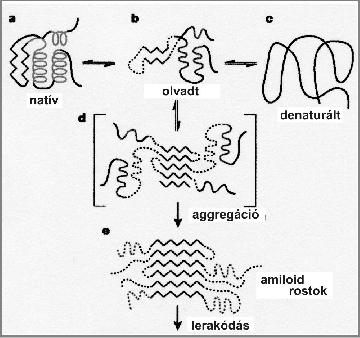
2. ábra. Az amiloid kialakulásának szerkezeti sémája
Az amiloid kialakulása az alábbi fő lépésekben történik. A fehérje funkcionális, natív állapotában jól definiált, stabil térszerkezettel rendelkezik (a). Ez a szerkezet véletlen külső behatások vagy mutáció hatására részlegesen felbomlik, egyfajta „olvadt” állapotba kerül (b). A fehérje leginkább ebben a köztes szerkezeti állapotban és nem a teljesen kitekeredett, denaturált állapotában (c) hajlamos amiloid képzésére. Az olvadt állapotban a polipeptidlánc bizonyos szakaszai a szomszédos fehérjével hidrogénhidak révén összekapcsolódnak (d). Ennek az átmeneti aggregált állapotnak a kialakulása általában hosszú ideig tart, és egy bizonyos méret alatt nem is megfordíthatatlan. Egy kritikus méret/stabilitás fölött azonban az aggregátum többé már nem alakul vissza, hanem további fehérjemolekulák befogásával egyre növekszik, és viszonylag szabályos, ismétlődő szerkezetű amiloid rostokká alakul (e). Ezek a rostok a szervezet különböző pontjain (a tárgyalt neurodegeneratív betegségek esetében az agyban) lerakódnak, és toxikusságuk révén elpusztítják a sejteket.
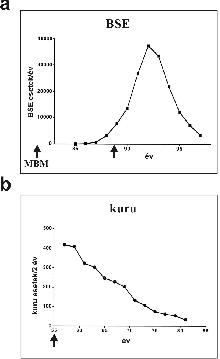
3. ábra. A BSE és kuru terjedésének, illetve visszahúzódásának időbeli lefolyása
A szarvasmarha szivacsos agyvelőgyulladás (BSE) és az emberi kuru (nevetőhalál) esetében is a járvány lassú terjedése, majd a megelőző beavatkozást követő lassú visszahúzódása volt megfigyelhető, ami a „lassú vírus” hipotézist látszott alátámasztani. A BSE esetében a betegséget az állattáp újfajta elkészítési módja (meat-and bone meal, MBM) indította útjára a múlt század hetvenes éveiben, a járvány visszafordulása pedig az MBM-táp beszüntetése (1988, második nyíl) után mintegy 4 évvel következett be (a). A kuru esetében a járvány felfutására nincsenek statisztikai adataink, a rituális kannibalizmus megszűnése (kb. 1955, nyíl) után azonban az új esetek számának lassú csökkenése volt megfigyelhető (b). Mindkét eset jól demonstrálja a késleltetést, ami a hosszú inkubációs időnek tudható be.

Friss hozzászólások
7 év 50 hét
10 év 23 hét
10 év 27 hét
10 év 27 hét
10 év 28 hét
10 év 29 hét
10 év 29 hét
10 év 31 hét
10 év 31 hét
10 év 32 hét